Click HERE for updates to this paper!
Andrew James Parker
Dept. Biomedical Science,
University of Sheffield,
Sheffield, England
Updated: June 2000
|
 |
Click Here
For Printer Friendly Version
|
Generic Name :
Temozolomide
USA Brand Name:
Temodar®
Other Names:
Temodal , Methazolastone, CCRG81045, SCH52365, NSC362856, M&B 39831
Classification:
Oral cytotoxic agent.
BT Dosage:
150-200mg/m2/day [Adult] Total dose: 750-1000mg/m2/cycle
60 -100mg/day [Children] Total dose: 900-1075mg/m2/cycle
Delivery: Oral tablet
Schedule: Five consecutive days, repeated every 28 days. (other schedules are under investigation).
Side Effects:
Possibility of nausea and vomiting, controllable by anti-emetics.
Less frequently: Headaches, rash, diarrhoea, constipation, alopecia,
high blood sugar, myelosuppression and elevated liver function tests.
Availability:
Temodar is now available by prescription in the USA.
Click here for availability in other countries.
Introduction
Temozolomide, or temodar, is the result of almost forty years of chemical evolution. It is derived from a range of drugs developed principally for antitumor activity in melanoma but has shown to have activity with other cancers. Amongst these, temodar has been noticeably effective (compared to other chemotherapeutic agents) in the treatment of glioma. Results have been particularly encouraging with astrocytomas. This report lists many of the recently published articles on temodar and attempts to set them into the context of the development of temodar.
Temodar was granted orphan status (marketing rights) in 1998
(167)
. In the same year, the European Medicines Evaluation Agency approved use of Temodar for progressive or recurrent glioblastoma multiforme after standard therapy then in 1999 for treatment-resistant anaplastic astrocytoma. In 1999, the US FDA gave approval of temodar for treatment-resistant anaplastic astrocytoma. This was a significant benchmark because the FDA has approved of only two other drugs for brain tumor treatment since the 1960s. The results of clinical trials are beginning to be collated
(154)
and reviewed
(138,
165).
A recent meeting of the American Society of Clinical Oncology (May 2000) presented over twenty abstracts focused on temodar. Given that there are currently in excess of thirty chemotherapeutic agents being used or tested in such cases, what attributes make this methylating agent worthy of such distinction? This short essay will attempt to summarise the emergence of temodar as a noteworthy treatment of primary brain tumors.
A detailed review of the development, chemistry and progression to clinical trials of temodar has been published
(1)
and other papers
(2,
3,
173,
181)
and reviews
(4,
5,
6,
7,
177,
180)
have examined the position of Temodar as a relatively new anti-tumor agent. The journey from formula to formulation is usually an arduous one and the precursors of temodar can be traced back to a group of compounds called triazines, which were developed in the 1960s. The progressive refinement of these molecules involving dacarbazine (DTIC) in the 1970s, and tetrazinones and mitozolomide in the 1980s serves as a paradigm for drug development, one that has been chronicled and likened to evolution
(8)
. Possibly the hardest (and rate-limiting) step in drug development is from concept to condenser but once synthesised there are a number of stages which have to be followed: -
(a) Structural and chemical analysis
(b) Pharmacokinetics studied
(c) Mechanisms of action researched
(d) Toxicity studies
(e) Screening in cell culture and animals
(f) Progression to clinical trials:-
|
|
|
|
Clinical trials are usually performed in three phases:
Phase (i) examines delivery and dosage in a
small number of patients, (Less than 20).
Phase (ii) Determines if treatment works as expected
Normally in a larger group of patients , (20 - 50).
Phase (iii) Compares standard care with good treatments
from phase (ii). Larger numbers are involved and the trial is randomised.
|
This format seldom proceeds in such a linear fashion, but serves as an outline for study. |
The molecular formula of Temodar, C6H6N6O2, reveals little of its structure but the systematic name: 8-carbamoyl-3-methylidazo [5,l-d]- 1,2,3,5-tetrazin-4 (3H)-one gives an indication of its complexity. The syntheses of temodar
(9,
10,
11,
12)
structurally related molecule
(13,
14,
15)
and derivative
(16)
have been described. In pharmacology, an analogue denotes a substance that is similar in appearance or function to another but different in origin or development. Temodar is the methyl analogue of mitozolomide and both molecules belong to a group classed as imidazotetrazinones. The structure of these bicyclic antitumor agents has been reported
(17,
18,
19)
. Temodar is also an analogue of the anti-melanoma drug dacarbazine (DTIC) introduced into clinical practice in the 1970s. It was established that DTIC required metabolic activation to the triazene MTIC
(20)
. Temodar and mitozolomide also yield this active metabolite, MTIC, by decomposition in solution
(21,
22,
23)
. Because it is the producer of the active metabolite, temodar is termed a prodrug
(24)
. The subsequent metabolism of temodar, in vivo, from its first product MTIC to the final position of its constituent atoms has been fully described
(1)
. High performance reversed-phase liquid chromatography (HPLC) has been used to quantify temodar in plasma and urine of patients receiving temodar
(25)
. HPLC has also been used to analyse the biologically active degradation product MTIC in human
(26)
and animal
(156)
plasma. Accurate measurements of drugs in biological fluids are essential to determine the action of drugs within the body - their pharmacokinetics.
Pharmacokinetic studies of drugs include the routes and mechanisms of absorption, the rate at which a drugs action begins and the duration of the effect, the biotransformation of the substance in the body, and the effects and routes of excretion of the metabolites of the drug. These parameters are usually referred to as ADME factors; absorption, distribution, metabolism and excretion. Temodar is given as an oral tablet. The presence of food has minimal influence on the absorption of temodal
(146,
168)
. Although it degrades rapidly in alkali conditions it is stable under acid conditions, which means it can tolerate stomach acidity
(193)
. However, other administration routes have been studied
(27,
28)
. Additionally, Sparta pharmaceuticals have signed a licence agreement with Schering-Plough for the use of Spartajet with temodar. Spartajet is a drug delivery system that allows poorly water-soluble and water insoluble compounds to be given by injection. This may enable intravenous administration for patients requiring higher concentrations or delivery to localized areas (Sparta press release).
The distribution and pharmacokinetics of temodar have been probed by NMR
(27,
139)
and PET
(29)
. Studies on temodar metabolism
(30,
184)
have given values for its half-life (t½) - the time taken to decrease plasma concentration to half its initial value - ranging from 0.42h in human plasma
(30)
to approximately one
(27,
31)
to two hours
(184)
. Whereas t½ for the reactive metabolite MTIC has been estimated at between 88 min
(31)
and 1.9h
(26)
. Excretion of temodar has been shown to take place via the kidneys of mice and men
(32)
. Extensive studies have been undertaken to establish the most suitable dose for temodal
(142,
150,
163)
a delicate balance between maximising toxicity to the tumor whilst minimising toxicity to the patient. The maximum tolerated dose has been established as 1000mg/m2 over 5 days in each 28-day cycle
(114,
119,
120,
121)
although further studies have considered protracted daily dosing
(216,
217)
. Pharmacological studies extend to consider analogues of temodar
(33,
34,
35)
and interactions with other drugs
(36,
38,
39,
193)
. The synergistic effect (action potentiated by combination) of temodal has been studied with a number of other agents - fotemustine
(37)
, cisplatin
(151)
, BCNU
(152)
, and thioguanine
(158)
. Such studies can give insights as to the mechanism of action of a drug an area that has received considerable attention for temodar.
The mechanism of action of temodar is complex and involves more than one site of action. In certain cancer cells, temodar has been shown to have an inhibitory action on enzymes such as esterase
(40)
and glyoxalase
(41)
. However, the main cytotoxic (cell-killing) action of temodar is effected by its role as an alkylating (or methylating) agent
(42,
43,
44)
, specifically the alkylation of DNA. The active metabolite MTIC adds methyl residues to nucleotides in the DNA molecule. Temodar has been reported as not altering the methylation of genes such as c-myc and C-Ha-ras
(45)
, genes containing many methylated sequences. A number of studies suggest Temodar may block cellular replication by indirectly inhibiting DNA methylation, cells not blocked having hypomethylated DNA
(46,
47,
48)
. The majority of mechanistic studies have concluded that the antitumor activity of temodar is due to the methylation of DNA. However, there exist a number of natural repair mechanisms, which counteract such processes
(196)
. Damage by temodal is defended by three repair mechanisms:
- 06-alkylguanine-DNA alkyltransferase (ATase)
- DNA mismatch repair.
- Poly (ADP-ribose) polymerase.
The cytotoxic and mutagenic (causing genetic mutation) action of antitumor triazenes such as temodar have been attributed to their ability to form DNA adducts (a combination of chemicals with DNA) by the methylation in the O6 position of the purine base guanine to form O6-methylguanine
(49,
50,
51)
. Repair of these methyl adducts is mediated by the DNA repair enzyme O6-alkylguanine-DNA-alkyltransferase (ATase) which removes alkyl adducts from DNA in a single step mechanism without co-factors
(170)
. This action has been studied in human brain tumor cell lines
(200)
and prolonged administration of temodar may inactivate the mechanism
(215)
. Temodar has been shown to decrease the activity of ATase
(52,
53,
54,
55)
and inhibition of ATase enhances the action of Temodar
(56,
162)
. Conversely, high levels of ATase reduce the effect of temodar
(57,
58,
59,
60)
and high levels have been reported in human brain tumors
(61)
. Depletion of ATase potentiates (enhances) the action of BCNU
(54,
62,
63,
143)
and CCNU
(53)
. The effect of temodar is also potentiated by the presence of an ATase inactivating agent; O6- benzylguanine
(64,
65,
66,
67,
68,
69,
166,
172,
188)
this action has been reviewed
(70)
and studied in cancer cell lines
(171)
.
Replication of DNA is usually accurate but there are occasional errors of base pairings. This is usually corrected by the "proofreading" DNA polymerases. However, mismatched base pairs sporadically become incorporated into DNA. In normal cells such errors are corrected by the mismatch repair system (MRS), a group of seven proteins with the primary function of recognising and correcting mismatched base pairs within the DNA helix. When the O6-methylguanine produced by temodar is processed by a normal MRS this leads to cytotoxicity. Unfortunately, loss of DNA MRS has been observed in a variety of cancers that confer resistance to methylating agents such as temodar
(71,
72,
73,
74)
this process has been reviewed
(75,
182)
. Deficiencies in the MRS have been studied in conjunction with temodar in other cancers
(185,
189,
190)
. Abnormalities in MRS have been demonstrated in gbm cells
(76,
136)
but have been shown to be intact in human medulloblastoma
(77)
. Further screening of brain tumor classes is evidently required.
It has been shown that temodar causes adducts at the O6-position of guanine and that subsequent processing by an intact MRS leads to cytotoxicity. However, temodar can induce methyl adducts at other positions, N7-guanine and N3-adenine. Another repair mechanism, the base excision repair mechanism is involved in the correction of such lesions. This repair mechanism can handle a variety of structural DNA defects and in mammalian cells many enzymes are involved in the process. Adenosine diphosphate (ADP) ribosylation is involved in DNA excision repair and the enzyme poly (ADP-ribose) polymerase (PARP) is activated by DNA strand breaks and mediates cellular responses to DNA damage
(78,
79)
. Inhibition of PARP
(148,
164,
179,
187,
191,
201)
has been shown to potentiate the cytotoxic effects of N7-methylguanine and N3-methylguanine
(80,
81,
82,
83)
thus improving the action of temodar. Similarly, PARP has been shown to utilise nicotinamide adenine dinucleotide (NAD) and low cell levels of NAD have been shown to enhance the effect of temodar
(84).
The molecular mechanisms of temodar continue to be investigated but another factor that distinguishes temodar from other agents is its ability to cross the blood-brain barrier (BBB). The BBB is a protective mechanism that ensures the stability of the brain environment. Homeostasis, the maintenance of stable conditions is essential in the brain, particularly with respect to ionic concentrations involved in the control of neuron function. Thus the BBB serves to permit water, nutrients and lipid-soluble substances such as anaesthetics, nicotine, and alcohol whilst prohibiting pathogens (bacteria and viruses), metabolic waste, proteins, toxins and most drugs
(133)
. The action is one of retardation rather than complete exclusion and is effected by the relative impermeability of the brain capillaries, possibly in conjunction with glial cells
(132)
. Many publications describe the ability of temodar to cross the BBB but few offer citations to support this claim. There is a report of temodar entering mouse brain
(8)
and a study on mitozolomide delivered intraperitoneally into mice found a rapid and extensive distribution although the brain contained the lowest drug level compared to other tissues
(85)
. Indirect evidence for temodar crossing the BBB can be derived from a study which added an angiogenesis inhibitor (TNP-470) as well as temodar in a rat model of glioma
(86)
. TNP-470 caused a reduction of capillary permeability and there was evidence to suggest that it decreased the uptake of temodar by the tumor. This study highlights the caution that must be exercised with combinational chemotherapies. PET studies have revealed a greater distribution of temodar in glioma than in normal brain
(29)
and it has been shown to enter the CSF
(37)
but overall published supportive evidence is scant. It has been stated that the BBB breaks down around gliomas
(87)
but again this claim offers no supportive evidence. Even the existence of the BBB has been questioned and whether it can be blamed for the failure of many chemothrapeutic agents
(88)
. It would seem that further investigation into the physiology of the BBB is merited.
Having considered the structure, chemistry, pharmacology and mechanism of action of temodar, the next stage is to test its effectiveness. It would be irrational to test a novel cytotoxin on patients without discovering what dosage is required or whether any side effects outweigh benefits. Additionally, it is important to establish the schedule of administration of the drug since its pharmacokinetics, metabolism and clearance in vivo determine its potency. Consequently, toxicity and pathfinder experiments are performed in animals or increasingly in tissue culture. The technique of cell or tissue culture allows cells to be grown and maintained in physiological conditions and their response to drugs can be repeatedly monitored.
Clinical trials of mitozolomide revealed that it caused thrombocytopenia
(89)
, a reduction in platelets caused by damage to bone marrow. Although disappointing this lead to the selection of temodar due to its lower toxicity. Toxicity studies are perforce performed on animals or more commonly cell lines
(140)
but such studies are frequently criticised. Cell culture can be said to be unrepresentative of the living milieu since factors such as tissue interaction and blood flow are absent and apart from moral complaints, animal models are often claimed to be unlike human beings. Such is the burden of the research worker attempting to alleviate disease. One approach that minimises criticism is that of the xenograft, a technique that transplants human tumors into animal hosts and the mouse has proven to be particularly suitable for this method.
In mice, temodar has shown to have activity against lung cancer
(90)
by interfering with the action of protein kinase C, an enzyme active in brain tumor
(91)
. It has been used to treat neoplastic meningitis
(176)
and temodar has shown inhibition of tumor cell growth in a dose-dependent manner in models of colon adenocarcinoma
(92,
202)
, pediatric solid tumor
(141)
and melanoma xenografts
(93,
161,
162)
. One of these reports
(93)
also confirmed the importance of the schedule of temodar delivery. An improved response was obtained when the initial dose was divided into five equal fractions and given on consecutive days. This approach had been suggested by an earlier study
(30)
. Antitumor activity of temodar has been demonstrated in rodents bearing human brain tumor xenografts
(136,
194)
. Temodar has been shown to improve the action of BCNU
(94)
, was more effective than procarbazine
(95)
and works synergistically with O6-benzylguanine (O6-BG), the ATase inhibitor both in mice
(96)
and apes
(69)
. Uptake of temodar in a rat glioma model was diminished by the presence of an angiogenesis inhibitor
(86)
.
The action of temodar on tumor cell lines has been examined
(97,
98)
. The latter study evaluated the cytotoxic effect of temodar on a panel of human tumor cell lines and a satisfactory cytotoxic action was observed with breast, ovarian and non-small cell lung cancers. Activity was also recorded in other cancer cell lines including; renal cell carcinoma, colon cancer, melanoma, sarcoma and prostatic and pancreatic carcinomas. Studies have also been undertaken on brain tumor cell lines
(183)
. One report analysed the response of 14 human medulloblastoma and glioma derived cell lines to temodar and streptozotocin
(99)
. Although the cells responded similarly to these two agents, the action of O6-BG as an adjunct to treatment was found to be variable. A later study
(100)
combined temodar with X-irradiation in a human gbm cell line and also in human colorectal adenocarcinoma (Mawi) cells. It was found that the gbm cell line was more sensitive to temodar than the Mawi cells but significantly, the latter had far greater ATase activity, which reduces the effect of temodar. Other studies have reported a synergistic effect between temodar and other drugs with gbm cell lines
(130)
and have developed chemosensitivity assays to assess the potency of neoplastic agents
(147)
.
Since temodar exhibited cytotoxicity to a variety of human cancer cell lines
(98)
and had been shown to have low toxicity in screening experiments
(30)
, progression to clinical trials was logical and desirable. The efficacy of any drug in clinical trials becomes more apparent as increasing numbers are performed. In the interim, summation of trials is problematic. Each study will produce a range of responses that are often difficult to classify, e.g. some patients may report that a drug alleviates their symptoms but no clinical changes are observed. For the purposes of this essay, an examination of temodars action in trials will first examine the performance in patients with cancers other than brain. Then a brief chronological summary of published clinical trials of temodar in primary brain tumor will be presented.
During the 1990s, temodar has been the subject of clinical trials for the treatment of many types of cancer. A review of systemic therapy of T-cell lymphomas
(101)
recommended larger phase II trials for (inter alia) temodar. One such study in low grade non-Hodgkin's lymphoma only had a response in 1 of 18 patients
(102)
, however all these patients had received previous treatment which may have affected the response to temodar and further trials with different categories of lymphoma are indicated
(137,
192)
. Following good preclinical results with dacarbazine in leukemia, a group of workers in Italy have evaluated temodar for leukemia with results suggesting it may have therapeutic potential
(103,
104,
105)
. Although showing potential against many human tumor cell lines
(98)
and xenografts
(106)
, response to temodar in patients with pancreatic cancer
(107)
, renal cell carcinoma (108) and nasopharyngeal carcinoma
(109)
has been disappointing. However, it is considered to have potential in the treatment of sarcoma
(175,
220)
, breast cancer
(110,
111,
198)
and has shown antitumor activity in hepatocellular carcinoma
(112)
. In the development of temodar, it may be recalled that dacarbazine and mitozolomide were significant in its evolution. These molecules have shown activity against melanoma . Since temodar transforms to MTIC, the active agent of dacarbazine, its action in melanoma has also been investigated
(210)
. Antitumor activity has been observed in phase I
(113,
218)
phase II
(114,
149,
219,
222,
223,
224)
and phase III
(160,
197,
199)
clinical trials. Measurement of ATase levels prior to treatment were not found to be predictive of temodar response
(115)
but reduction of ATase by O6-BG is advocated
(116,
161)
. Temodar has been applied in dural melanoma
(155)
but did not produce a response in uveal melanoma
(117)
. Reviews of the chemotherapy of melanoma
(118,
195)
suggest that temodar shows promise, especially in cases with brain metastases. Temodar has been shown to prevent brain metastases in metastatic malignant melanoma
(221,
225)
. The consensus opinion is that trials of temodar should focus on melanoma and glioma.
To date, the greatest potential of temodar has been manifest in the treatment of primary brain tumors. This intractable cancer has proven resistant to many promising avenues of treatment in the past and success is long overdue. It should be stressed that temodars efficacy is still under review. Phase I
(121,
128,
174)
and II
(76,
122,
157,
162)
trials have taken place, with progression to randomised phase III trials being advocated.
(87)
and initiated. The present article will briefly outline results of clinical trials of temodar on brain tumors published over the past eight years.
Phase I trials started in England in 1987
(119)
examining the response to single dose (50 - 2000 mg/m2) given intravenously and oral dose repeated over five days (total: 750-1,200mg/m2). Although this trial only generated two partial responses in patients with recurrent high-grade glioma, it established the importance of the dosing schedule. Using that regimen, a later study
(120)
found major improvement in computer tomography (CT) scan in 5/10 patients with astrocytomas and major clinical improvement in a further patient. Reduction in size of the CT scan was observed in 4/7 patients with high-grade astrocytomas given 2-3 courses of temodar prior to radiotherapy. A phase I study investigating toxicity
(135)
found that grade 4 thrombocytopenia was the dose-limiting toxicity in 2 of 3 patients at a dosage of 250mg/m2 for five days. Continuing work at the Charing Cross Hospital in London was described in 1996
(121)
in a study of 75 patients with malignant glioma. The overall objective response rate was 27% but the responses were of short duration. The authors suggested that quality-of-life evaluation should be included in such studies, even if a drug does not cure, any alleviation of symptoms must be considered. A phase II trial of 103 patients with recurrent high-grade gliomas
(122)
achieved an objective response in 11% and a further 47% had stable disease. Response rates were similar for aa (grade III) and gbm (grade IV). As in previous studies, myelosuppression was the major toxicity.
Investigation into alternative drug schedules reported in an abstract in 1997
(123)
found an objective response in 7 of 15 patients (47%) receiving 75mg/m2/day over 6 or 7 weeks, thus permitting delivery of a total dose approximately double the conventional one and suggesting further investigations into alternative scheduling. In the same year, an American phase I trial
(124)
found a difference in response between patients with or without prior exposure to nitrosurea (NU). There were two complete responses (one glioma and one melanoma) in patients without NU. Differences between previously treated and chemotherapy naive subjects has been discussed
(102)
. The study recommended increasing dosage to 225mg/m2/day for 5 days for patients previously treated with NU.
Further abstracts in 1997 reported pharmacokinetics of temodar as studied by PET scans
(125)
and as influenced by cisplatin
(126)
. The latter did not generate any significant cross-reaction. Two more American studies concluded in favor of temodar. The first, of thirty three patients with newly diagnosed gbm and five with aa, conducted at Duke University
(76)
, found 3 complete responses, 14 partial responses and 4 patients with stable disease. The second was a phase II multicenter study performed over 19 months at 32 centers evaluating 161 patients with AA
(127)
. The overall response rate (complete and partial) was 42 % and as with other studies, toxicity was found to be mild with less than 6% experiencing some myelosuppression.
The Charing Cross Group in 1998
(128)
published additional studies into alternative schedules. Twenty-four patients (17 gliomas) received daily doses of temodar starting at 50mg/m2/day increasing to 100mg/m2/day for up to 7 weeks. Of the glioma patients, 41% demonstrated tumor responses. This new regimen was recommended for further studies.
Two phase I studies of paediatric patients have been published. The UK Childrens Cancer Study Group
(131)
evaluated 16 patients without prior NU or craniospinal irradiation (CSI) and used on 5 consecutive days doses ranging between 500 and 1200mg/m2 per cycle. Their recommended dose was 1000mg/m2 per cycle. Responses were noted in 2 out of 5 patients and one patient had stable disease. The report from the Childrens Cancer Group in Washington
(129)
evaluated a range of doses in 27 patients. The study recommended a maximum tolerated dose of temodar in children of 215mg/m2 for those without prior craniospinal irradiation (CSI) and 180 mg/m2/day for 5 days for those with prior CSI. Ten patients had stable disease, 3 had partial response and one had complete response persisting over two years of follow up.
At an international meeting in Versailles on Sept. 15th 1998, results of a phase II/III clinical study were presented. Dr Yung reported on an evaluation of temodar in 225 gbm patients, randomised to receive either temodar or procarbazine. On all parameters reported in the Schering Plough press release, temodar performed better than procarbazine. Six-month survival being 60% in the temodar group compared to 48% in the patients receiving procarbazine. At the same conference, Dr. Brada reported on a phase II study in 162 patients with aa at first relapse. Progression free survival at six months was 46 % and at twelve months 24 %. The overall objective response rate was 35 % and stable disease occurred in 27% of patients. A large proportion of patients had improved quality-of-life scores.
Trials continue to investigate the pharmacokinetics of temodar
(163,
168)
and to produce encouraging results particularly with anaplastic astrocytoma
(157)
. The combination of temodar with other agents is still being investigated
(170,
174,
178,
227)
. The number of trials being reported allows for comparative study
(138,
165)
but there are fundamental problems associated with clinical trials. Every patient is an individual for whom the physician will strive to effect the optimal treatment. It is therefore problematic to achieve a "standard" procedure. Any patient group will consist of mixed gender with a wide range of age, prior treatments, disease type and severity. The guidelines for selection into clinical trials are strict, because any selection bias (choosing patients most likely to respond) would influence the outcome. Similarly, analysis of the results of trials is fraught with obstacles. The performance indices are not standardized and different researchers use differing measures. This variation is long overdue for international alignment. It is sometimes impossible to reconcile what one study means by "Complete Response -CR" or "Partial Response - PR" with that of another group. Some groups present CR and PR as one statistic. Sometimes different diseases are analysed together (e.g. glioma and melanoma;
124,
142,
168)
or in some cases the type of cancer is not specified
(126)
. Some reports concentrate on quality of life
(134)
but there are as many different ways this can be measured as there are reports.
|
In an attempt to present the results of recent and landmark clinical trials in an assimilable form, the following table has been constructed.
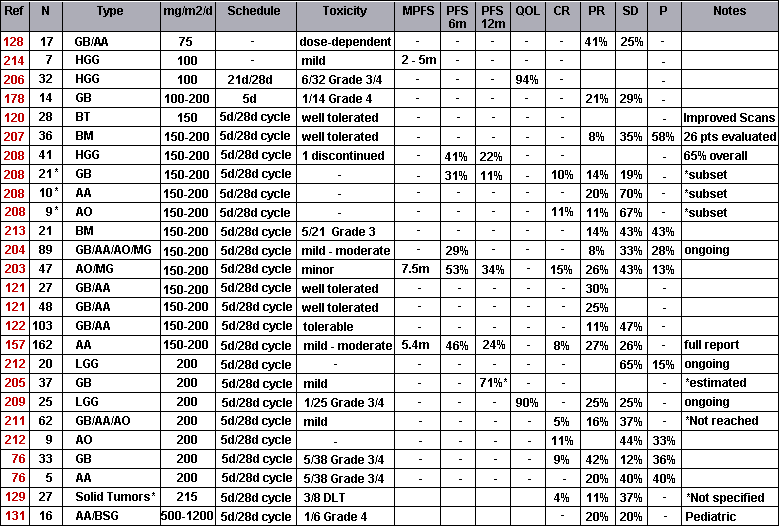
| Abbreviations: |
|
| Type: |
AA = Anaplastic Astrocytoma
AO = Anaplastic Oligodendroglioma
BM = Brain Metastases
BSG = Brain Stem Glioma
BT = Brain Tumor
|
|
GB = Glioblastoma
HGG = High Grade Glioma
LGG = Low Grade Glioma
MG = Mixed Glioma
|
|
Time:
d = day; w = weeks; m = months
|
|
|
Parameter:
|
MPFS = Median Progression Free Survival
PFS = Progression Free Survival
QOL = Quality of Life
CR = Complete Response
|
|
PR = Partial Response
SD = Stable Disease
P = Progression
|
|
Misc:
Pts = Patients DLT = Dose Limiting Toxicity
|
The table presents (from left to right): The reference for the trial; Number of Patients; Dosage; Schedule; Toxicity; followed by the various reported parameters. All percentages have been approximated to the nearest integer. Most of these studies are based on chemotherapy following surgery and radiotherapy (one as an adjunct to radiotherapy
(205)
. A number of points become apparent. The toxicity is generally low with only one patient discontinuing treatment due to adverse reaction. The grades reported refer to incidences of toxicity over a range of possible side effects (e.g. nausea, myelosuppression, and neutropenia) - mostly haematologic. Progression free survival extends to an estimated 71% at 12 months
(205)
. Complete Response, Partial Response, and Stable Disease are approximately 10 %, 20% and 40%, respectively.
What is hard to discern is the variation in response amongst glioma sufferers. One study reports subgroups
(178)
. Is it possible to detect those patients who will best respond to temodar? This is clearly one aspect of brain tumor therapy that urgently requires attention - the matching of tumor susceptibility to the appropriate chemotherapy. Evidence for this may come from chemosensitivity assays
(147,
153)
or from noninvasive techniques such as NMR
(139,
159)
. This has already been achieved with another chemotherapeutic
agent
(226)
. Is it pos sible that our nomenclature and histology of brain tumors is still in its infancy and that further classification will facilitate treatment?
In summary, temodar is not a novel drug but the result of some forty years refinement
(1)
and may not be the end product of the process
(144,
145,
169,
186)
. It is a prodrug
(24)
with excellent oral bioavailability and biodistribution that crosses the blood brain barrier
(37,
85)
and decomposes to the active metabolite MTIC
(21)
in alkaline conditions
(193)
. Gliomas are reported to have an alkaline environment
(132,
133)
. The mechanism of action, although not fully understood, has been extensively analysed enabling biochemical enhancement
(56,
70,
80)
of its cytotoxicity. As a chemotherapeutic agent it has low toxicity with myelosuppression presenting the major challenge
(122,
127)
. Although many studies have been performed to establish optimum dosage
(119)
and schedule
(128)
, alternative regimens may yet be investigated
(150,
163,
168)
. Interactions with other drugs merits further attention both in potentiation
(53,
94,
151,
152,
227)
and interference
(37,
39,
85)
. Additionally, diminishment of temodar's action by other chemotherapeutic agents
(124)
and angiogenesis inhibitors
(86)
needs further study. The gulf between in vitro and in vivo studies is highlighted by demonstrable cytoxicity against human cancer cell lines
(97,
98,
164,
183)
failing to be matched in clinical trials
(107,
108,
109)
. Early clinical trials have shown to be of benefit both in anaplastic astrocytoma and glioblastoma multiforme, not only in reduction of tumor size but also in extended progression free survival and improved quality of life
(134)
. This essay may have indicated some aspects of the difficulties associated with the process of drug development. The rigours of the scientific method sometimes appear to hinder progress but they ensure that precious resources are not misdirected in the pursuit of drugs of minimal or irreproducible benefit. The long journey from test tubes in the 1960s to tablets in the 1990s has indeed been arduous but when a trial reports on a child with complete response to glioma lasting over 2 years after administration of temodar
(129)
, the first reaction is simply appreciation. Ultimately the clinical statistics will reveal the full story.
|
|
|